
���A����Clustalx��ʹ�÷���
�r�g��2019-09-20 11:49:57 ���ߣ��o�� �g�[����63
����С���͞��Ҏ������A����Clustalx��ʹ�÷�����CLUSTALX����CLUSTAL�������бȌ������Windows�汾��
- �M�����d

- ClustalX 1.83 �ٷ���
����502 KB
���ڣ�2019/9/20 11:49:57
�h����WinXP,Win7,
Clustal��һ�N���Ýu������progressive alignment���M�ж��l���бȌ���ܛ�������Ȍ��������ЃɃ����^�������x��ꇣ���������֮�g�Ƀ��Pϵ��Ȼ��������x���Ӌ��a��ϵ�y�M��ָ���䣻Ȼ��Ķ��l�����������ƣ����x���ڣ��ăɗl�����_ʼ�Ȍ������ո����������������ϵ�λ�ã��ɽ����h�����������������������Ȍ���ֱ���������ж��������γ���K�ıȌ��Y����ֹ��Figure 1����

clustal �㷨��
Clustalܛ���Ѓɂ��汾������clustalw���������е���ʽ��DOS/Linux���\�еģ� Clustalx�ǿ�ҕ������ij�����window��X�\�У��҂�����W��Clustalx��ʹ�á�
1 ���bclustalx��
���dclustalxܛ��������Ĭ�J���b���Լ�����X�ϡ�
2 �ʂ�Ҫ�Ȍ������У�
���Ϲ��n��������ͬԴ����fasta�ļ���ȫ��ճ�N��һ���ı��ļ��У����еĵ����|���д�����һ���ı��ļ���

���A����Clustalx��ʹ�÷����D��
3 �d������
�c���_ʼ������clustalX2��clustalX2��
�c���ˆ�File���x��Load Sequence-�x����������ļ����c���_��
ע�⣺ClustalX����o���R�e�h�֡�����λ���ļ��A������my document��
�d�����к�����ȴ�������fasta��ʽ���еĘ��R̖��ȡ�����е�һ��“>”����ַ���
4 �Ȍ��������O�ã�
�Ȍ�ǰ��Ҫ�O�Ãɗl���бȌ��ą����Ͷ��l���бȌ��ą�����
a.�ɗl���бȌ��ą���
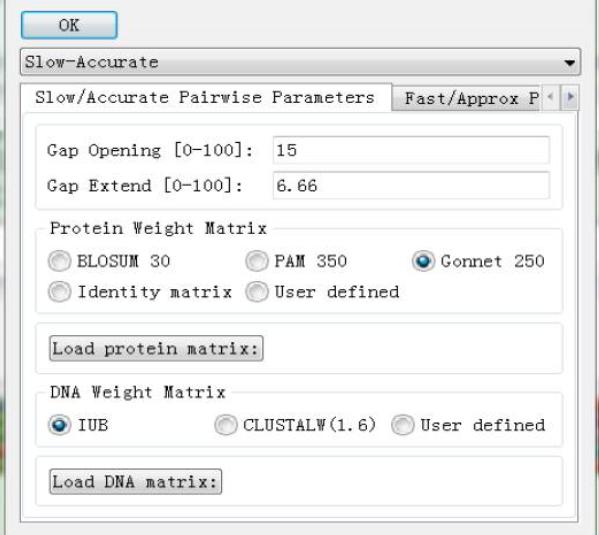
�c��Alilgnment�ˆΣ��x��Alignment Parameters�����x��Pairwise Alignment Parameters����Figure 3�����ȿ����x��Ȍ���Ч������slow/accurate ߀��fast/approximate����һ�Nģʽ���õ��DŽӑBҎ���㷨�M�бȌ��ģ��ڶ��Nģʽ���õ��dž��lʽ���㷨���������зdz��L��һ����õ�һ�Nģʽ�������x���λ�P��ϵ�y��DNA���|��Q��ꇣ�Ҳ�����Լ��ς�ij����Q����M�бȌ���
b.���l���бȌ�����
�c��Alilgnment�ˆΣ��x��Alignment Parameters�����x��Multiple Alignment Parameters����Figure4.
Delay divergent sequence��ָ���ɗl���еIJ����ij��ֵ���ٷֱȣ��r���@�ɗl���еıȌ������t�M�У������ȱȌ��������У��������ƶȲ���ߵ����У���Щ�r���M�бȌ������뵽��K�Ķ��l���бȌ��Y���rҲҪ�tЩ��DNA transition Weight����0�ĕr�����D�Q�����e�䣨mismatch������������1�ĕr���D�Q��ƥ��ͬ�ȿ����������c�Ȍ������в�^��r��DNA transition Weight��ԓ�x���СЩ���ӽ�0����������c�Ȍ������в�^С�r��DNA transition Weight���x��Ĵ�Щ���ӽ�1����
���A����Clustalx��ʹ�÷����D��
5 �O��ݔ����ʽ��
�c��Alignment�ˆΣ��x��Output Format Options�������Figure 5��
Ĭ�J����ݔ��clustal format�������Ҫ������ʽ�����ڏ��x�������PHYLIP��ʽ������PHYLIPܛ���M�н���r����Ҫݔ��ĸ�ʽ
6 �M�бȌ���
�c��Alignment�ˆΣ��x��Do Complete Alignment.�˕r���Fһ����Ԓ����ʾ�Ȍ��Y�������λ�ã���һ���x���˶��ٷNݔ����ʽ���@�����Ҫ�o�����ق��ļ���·�����x������cOK���ɡ�
ClustalX 1.83 �ٷ���
- ܛ�����|������ܛ��
- �ڙʽ�����M��
- ܛ���Z�ԣ�Ӣ��
- ܛ����С��502 KB
- ���d����615 ��
- ���r�g��2019/9/17 13:29:54
- �\��ƽ�_��WinXP,Win7,...
- ܛ��������CulsTAL X��CulsSTW�����бȌ������Windows�ӿڡ����ṩ����... [�������d]
���Pܛ��
502 KB / 2019/9/20
- ��֦FM����6���������µļm��?
- 10000��˾�C���V�㣺�ε�Ҳ���ڶ��˶��ɣ���...
- ը��С����淨Ԕ�� ����С���ɅR��
- ������APP�ϏV������Ͷ�M����������?
- �ְ��ֽ�������á����۲顱��һ�r�g�l���...
- B612�������o��ͯԒ���X����������
- �y�̾W��ô���̓r�CƱ �y�̓��̓r�CƱ����
- ֪����߅��С���ɣ������@Щ������p������...
- ��������I�Ϳ��ٌ��Ҵ�
- ���FС���ɡ���W���ˆ�?
- ����qq�w܇�a���������w�Y����
- ��ʲôҪ�x��mplayer������
- �Ԍ��ȃrܛ�����ݻ�ُ�����ֵ�ʹ�÷���
- ʲô��akelPad��С�����㿴����
- �\ՄDOTA2�����ӡ� ��������Ɖ��Α�ƽ��...
- �Ʒ�����ƽ�_���d�������}��Q�
- �V���첥���ò����ļ���
- �ı���x���Ŀ��?6���ı���x�����^
- tq�ͷ�ϵ�y�aƷ��B
- PPVOD������ʲô?PPVOD����ʲô����?
����������Xܛ��
 Ӣ����
Ӣ���� ���ߘsҫ
���ߘsҫ ���LӰ��
���LӰ�� �ṷ����
�ṷ���� �vӍQQ
�vӍQQ 360�g�[��
360�g�[�� 360��ȫ�lʿ
360��ȫ�lʿ 360����
360���� Ѹ��
Ѹ�� �ѹ�ݔ�뷨
�ѹ�ݔ�뷨 ����
���� �Ӿ��`
�Ӿ��` ���D����
���D���� WinRAR
WinRAR WPS Office
WPS Office ��ɽ����ͨ
��ɽ����ͨ


